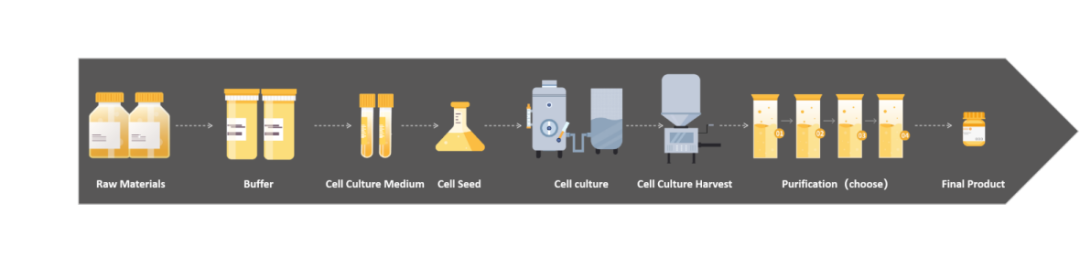
近些年,随着生物医药迅猛发展、疫情下细胞基因治疗以及mRNA疫苗的相继涌现,如何把控生物制品安全可靠,成为各国法规政府重视以及监管的要点。支原体是一种比较常见但通常难以去除的污染类型,对于涉及细胞培养的生物制品工艺过程,法规要求“必须确保无支原体污染”。
-
2020版中国药典三部《生物制品生产检定用动物细胞基质制备及质量控制》中提出对于生产用细胞,需要对主细胞库(MCB)、工作细胞库(WCB)、生产终末细胞(EOPC)进行支原体检查。
-
《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》中建议在关键时间点对适合的中间样品开展支原体等安全性相关检测或采取相关的措施加以控制,并且支原体同样需要作为终产品的放行检项。
-
FDA,Guidance for Industry:Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications,中提出对于原材料、病毒种子、未加工的收获液等都需要进行支原体控制。
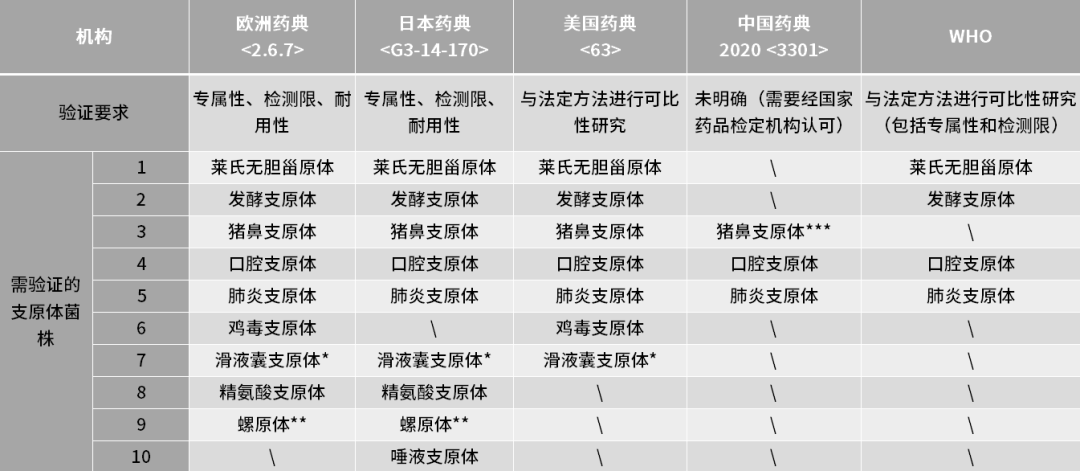
在快速检测方法(NAT,核酸扩增技术)出现之前,传统的培养法和指示细胞法因其检测周期长或灵敏度问题,致使企业生产或产品放行周期拉长或者需要基于风险评估对细胞原料进行条件放行或者寻求其他替代方法。而随着细胞基因治疗的发展,行业内对支原体检测时效性和灵敏度要求越来越高,短的货架期不足以支撑如此长的检测周期,因此NAT方法从众多支原体检测方法中脱颖而出。 目前欧洲药典(EP)<2.6.7>,日本药典(JP)以及美国药典(USP)<63>都已收录NAT方法作为支原体检测方法,但需要对该方法进行验证以及与传统方法进行对比其检测灵敏度不低于传统方法,才可以使用。2020版中国药典(ChP)虽然并未收录NAT法作为支原体检测方法,但其中也提到了“也可采用经国家药品检定机构认可的其他方法”。2022年5月,国家药监局药审中心发布的《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》中提到“当检验样品量有限,或需要快速放行等特殊情况下,如药典方法无法满足,可考虑开发新型的无菌和支原体的检测方法进行放行检测,但是新型检测方法应经过充分验证”。所以NAT法有望作为能够支持快速支原体放行检测的方法,被越来越多的原材料供应商、以及ATMP企业所使用。 对于如何进行NAT方法的验证,目前欧洲药典<2.6.7>和日本药典里都详细介绍,其内容基本一致。中检院也发表了文章《支原体检查的核酸检测方法及方法学验证的思考》,以期对我国支原体核酸扩增法检测的研发者及使用者提供借鉴。支原体NAT法须进行专属性(Specificity)、检测限(Detection Limit)和耐用性(Robustness)验证。如果使用商业化试剂盒进行支原体检测,则可以基于供应商的完整试剂盒性能验证报告再结合本身实验室环境和样品类型进行合适的方法适用性验证。 专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。EP中提到需要在验证中关注与系统发育关系比较近的其他细菌种属之间的交叉反应,如革兰氏阳性菌包括梭菌属Clostridium、乳杆菌属Lactobacillus和链球菌属Streptococcus。当然也需要关注宿主细胞和实验操作环境中比较常见的如人源DNA污染类型。 检测限系指试样中被测物能被检测出的最低量。检测限仅作为限度试验指标和定性鉴别的依据,不需要被准确定量为一个准确值。EP中要求每种支原体每个稀释浓度都需要有24次检测数据,可以在不同天做3次独立的10倍系列梯度稀释,每个稀释梯度做8次重复检测;或者可以选择不同天做4次独立的10倍系列梯度稀释,每个稀释梯度做6次重复检测。检测阳性率需要满足95%以上可以作为检测限。 对需要进行检测限确认的支原体类型,试剂盒厂家需尽可能多的覆盖法规药典中要求的支原体类型,并需要对每种支原体的检测限进行摸索。而对于生物制品企业来说,通常需要根据实际使用的原辅料来源进行选择。另外建议把人源支原体类型考虑进去。
-
如果在生产过程中使用或接触禽细胞或材料,应进行滑液囊支原体的检测限验证;
-
如果在生产过程中使用或接触昆虫或植物材料,应进行螺原体的检测限验证;
-
猪鼻支原体在支原体污染样本中占比非常高,受到中国食品药品检定研究院的关注。
相比于欧洲药典,日本药典中对于需验证检测限的支原体中,并未提到鸡毒支原体而是增加了唾液支原体。 耐用性系指在测定条件有小的变动时,测定结果不受影响的承受程度,为所建立的方法用于常规检验提供依据。对于耐用性的评估,需要关注支原体检测方法对于检测试剂中MgCl2、引物和dNTP的浓度变化,核酸提取试剂盒或提取步骤的改变以及使用不同核酸扩增仪的耐受程度。
可比研究(Comparability Study)
如果要使用NAT替代药典方法,则需要通过对比确认NAT法能够替代培养法或者指示细胞培养法,通常需要考虑方法的检测限,同时也需要把专属性(如覆盖的支原体范围以及可能的假阳性)考虑进去。对于检测限的对比,需满足“检测限达到10 CFU/mL,可以替代培养法;检测限达到100 CFU/mL,可以替代指示细胞培养法”。 1. 使用相同验证支原体菌株,同时进行NAT法和药典方法的同步实验,确认检测限; 2. 使用NAT法验证结果与之前的药典方法验证结果进行对比,但对于2种方法中使用的支原体标准品的确认文件需要仔细记录。
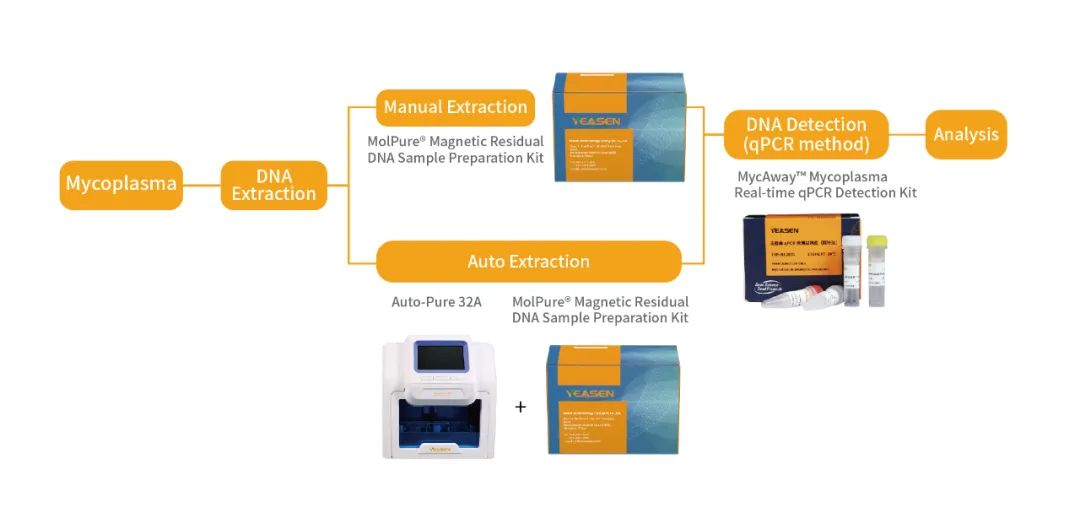
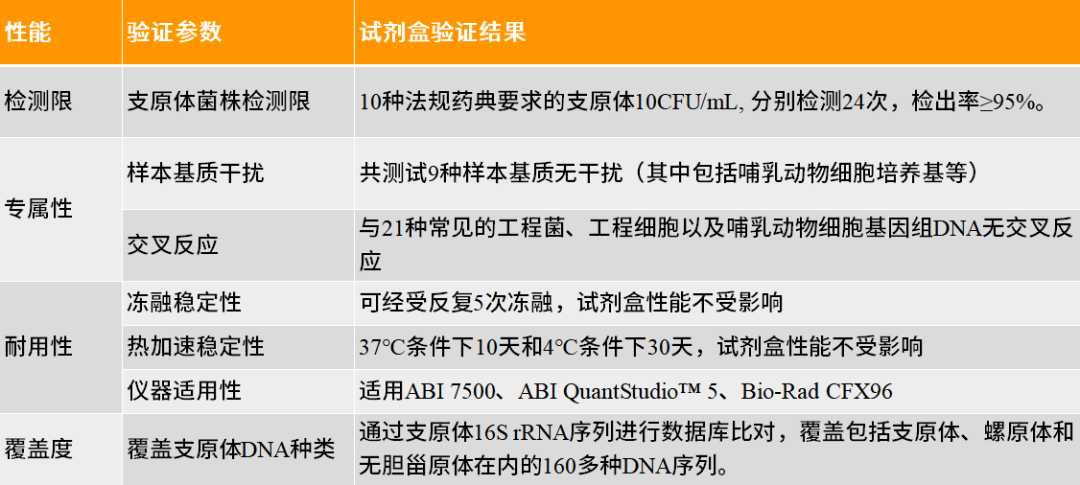
翌圣生物MycAway™ 支原体qPCR检测试剂盒(探针法)是基于NAT(nucleic acid amplification techniques)的一种快速定性检测生产原料、细胞库、病毒种子、病毒或细胞收获液、治疗用细胞中潜在支原体污染的产品。该试剂盒基于定量PCR技术,使用Taqman荧光探针定性检测待测样本中支原体DNA,可覆盖160多种支原体DNA序列;并且严格按照EP 2.6.7和JP G3支原体检测相关指南和要求进行验证,具备灵敏度高、特异性好、安全性好等特点。 它能够与MolPure® 磁珠法残留DNA样本前处理试剂盒搭配使用,可以通过手动提取或者使用Auto-Pure 32A全自动核酸提取仪自动提取样本核酸。样本经过前处理去除干扰杂质获得纯化的支原体DNA后,通过qPCR收集探针的荧光信号,从而对检测结果进行判定。
翌圣为您提供MolPure®磁珠法残留 DNA 样本前处理试剂盒(货号18461ES25)和MycAway™支原体qPCR检测试剂盒(探针法)试用装,每个客户限申请1套(即2种试剂盒各1个),总量限购40个(最终解释权归翌圣生物所有)。
扫码填写您的试用申请